intro-to-rnaseq-with-galaxy
Read Alignment
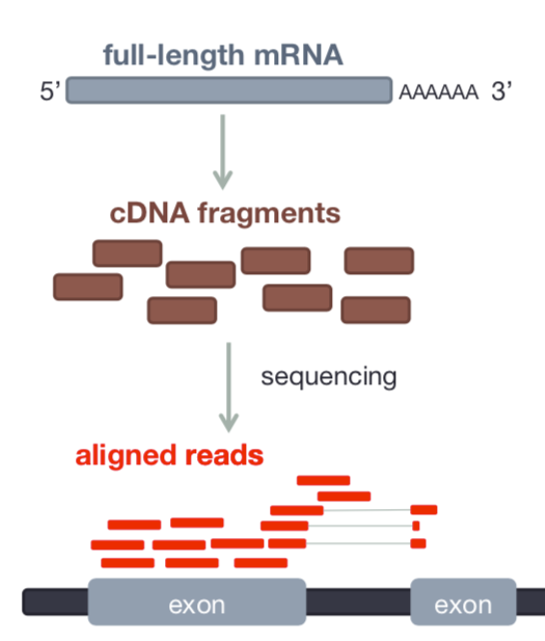
- RNAseq data originates from spliced mRNA (no introns)
- When aligning to the genome, our aligner must find a spliced alignment for reads
- We use a tool called STAR (Spliced Transcripts Alignment to a Reference) that has a exon-aware mapping algorithm.
SAM format
STAR produces a file in Sequence Alignment Map (SAM) format or the compressed version BAM.

Genome Annotation Standards
- STAR can use an annotation file gives the location and structure of genes in order to improve alignment in known splice junctions
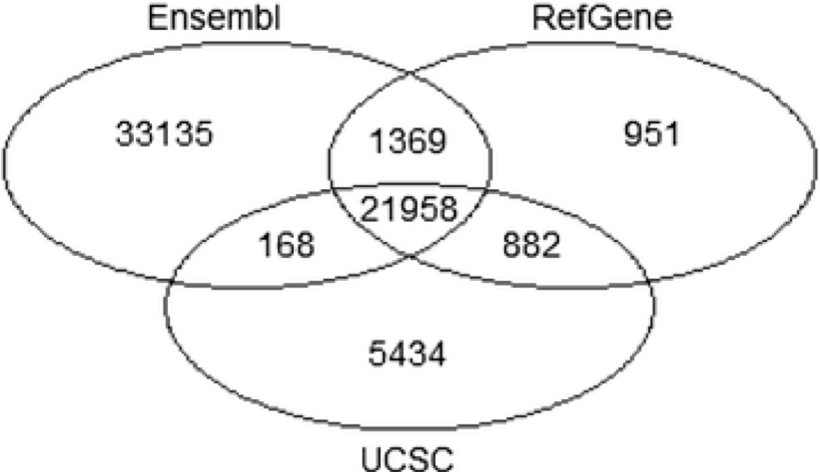
- Annotation is dynamic and there are at least three major sources of annotation
- The intersection among RefGene, UCSC, and Ensembl annotations shows high overlap. RefGene has the fewest unique genes, while more than 50% of genes in Ensembl are unique.
- Be consistent with your choice of annotation source!
GTF Gene Annotation
- In order to count genes, we need to know where they are located in the reference sequence
- STAR uses a Gene Transfer Format (GTF) file for gene annotation

Import a gene annotation file from a Data Library to be used for feature counting
- Click Shared Data on the top menu bar and select Data Libraries
- Select annotation_files
- Select the box next to hg38_genes.gtf and hg38_genes.bed.
- Click Export to History next to the Search bar and choose as Datasets
- Click Import to add the file to our current history
- Click
**Tufts Galaxy** in the top left to return to the homepage
Align the reads to the human genome using STAR aligner
- In the Tools panel search bar, type STAR
- Scroll down and select RNA STAR under RNA-seq
- Under RNA-Seq FASTQ/FASTA file click the
 and select the trimmed reads 42: Trim Galore! on collection 12: trimmed reads
and select the trimmed reads 42: Trim Galore! on collection 12: trimmed reads - STAR gives us the option of using a genome that includes a database of known splice junction locations or providing a gtf file so that STAR can create the database. We’ll select a reference genome on our server that does not include the splice junctions. Under Reference genome with or without an annotation select use genome reference without a built-in gene-model but provide gtf.
- Under Select reference genome select hg38.
- Under gene model select hg38_genes.gtf
- The final configuration should look like this:
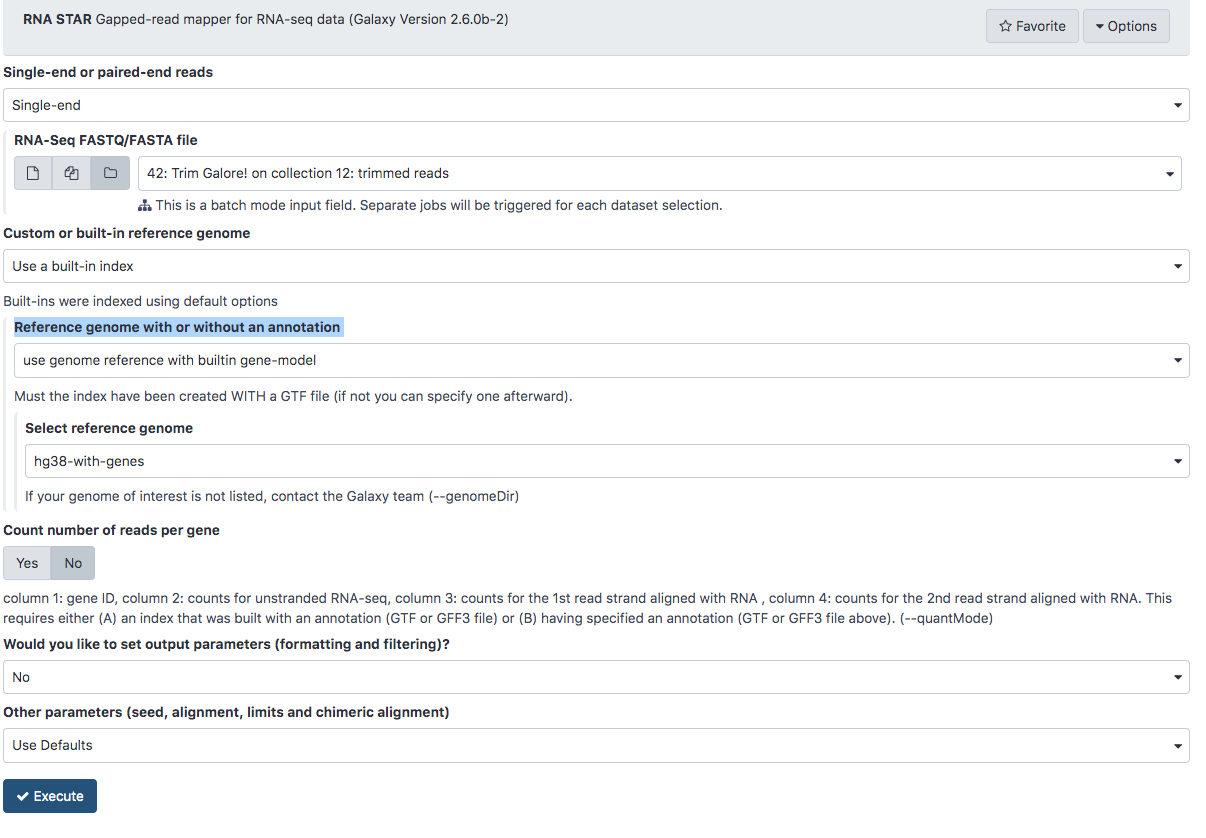
- Scroll down and click Execute
- The result will be three collections, giving the bam, splice junctions and log files for the alignments
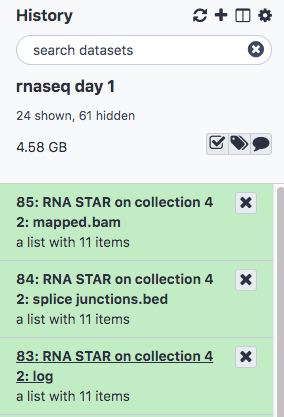
Run MultiQC on the STAR log files to check the result of the alignment
- Follow the steps from the previous section to run MultiQC except:
- Under Which tool was used generate logs? select STAR
- Under STAR log output click the and select the collection 83: RNA STAR on collection 42:log
- After the job finished, click the to view the webpage.
Question 5: In RNAseq, the percentages of uniquely aligned reads are typically lower than for DNAseq, due to the presence of unremoved ribosomal RNA. These are present in multiple copies throughout the genome and cause reads not to be mapped confidently. RNAseq is expected to be above 75% for an uncontaminated human sample. Is the "% Aligned" above 75% for these samples? You can optionally check to see which percentage of the reads align to the HIV genome by re-running STAR using the HIV genome and hiv annotation files located in Shared Data library called **
View bam file using JBrowse
- In the Tools panel search bar, scroll down to Graph/Display Data and select JBrowse genome browser
- Under Select a reference genome select hg38
Next we’ll add two Track groups, each with an annotation track
- Under Track Group click + Insert Track Group
- Click + Insert Annotation Track
- Select track type BAM Pileups and under BAM Track Data click the folder icon and select the list RNA STAR on collection: mapped.bam
- Scroll down and click Insert Annotation Track
- Select track type GFF/GFF3/BED Features and under GFF/GFF3/BED Track Data select hg38_genes.bed.
Finally, run the job:
- Scroll down and click Execute.
- Once the job is complete (green) click the eye icon to view the data.
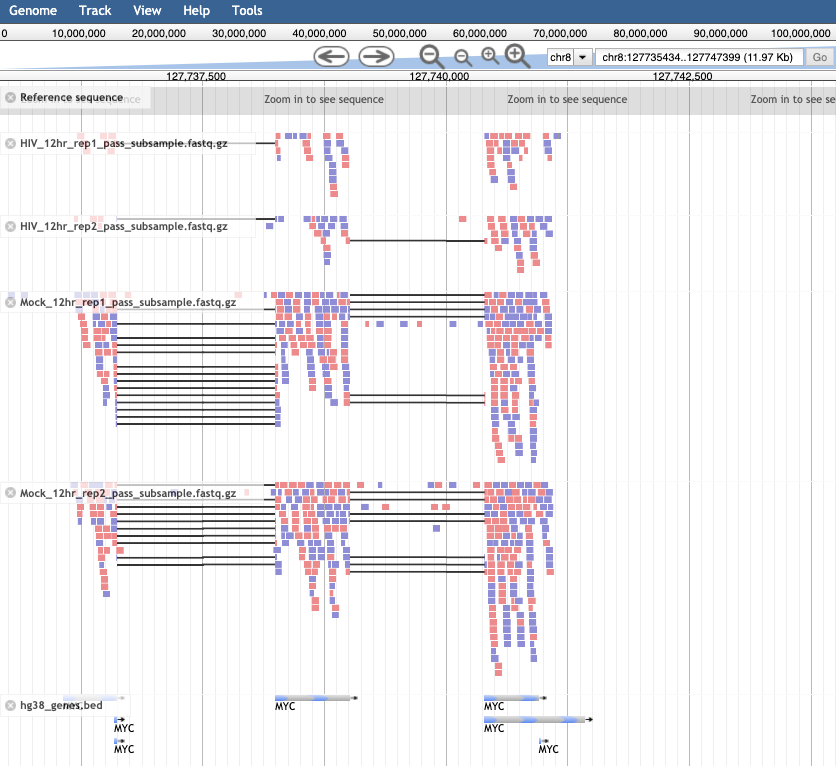
- In the Available Tracks panel select the HIV and Mock samples from 12 hr, as well as the bed file.
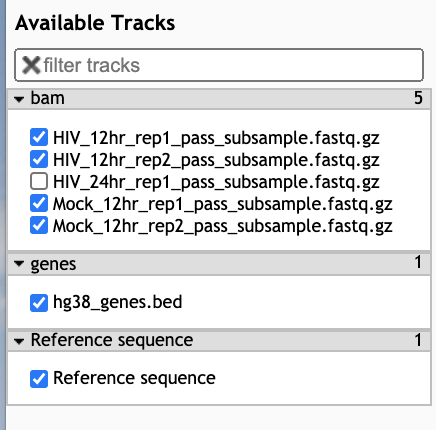
- We’ll zoom in on one gene MYC. To do this, click on the search bar to the left of the Go button and type
chr8:127735434-127742951. Note that you can’t search by gene name in this tool. - The bam tracks will show the reads that align to the region for each sample.
- The color will show whether the read aligns to the + or –strand and grey lines show splice regions where a read spans an intron. The gene track at the bottom called hg38_genes.bed will show 6 features of EGR1, by clicking on them you will be able to see the different feature types (exon, CDS, start_codon, stop_codon).