intro-to-rnaseq-with-galaxy
Gene Quantification
Counting reads for each gene
Our next step is to quantify the spliced reads that aligned to each gene in our GTF file For two non-overlapping, multiple-exon genes, our alignment may look like this:
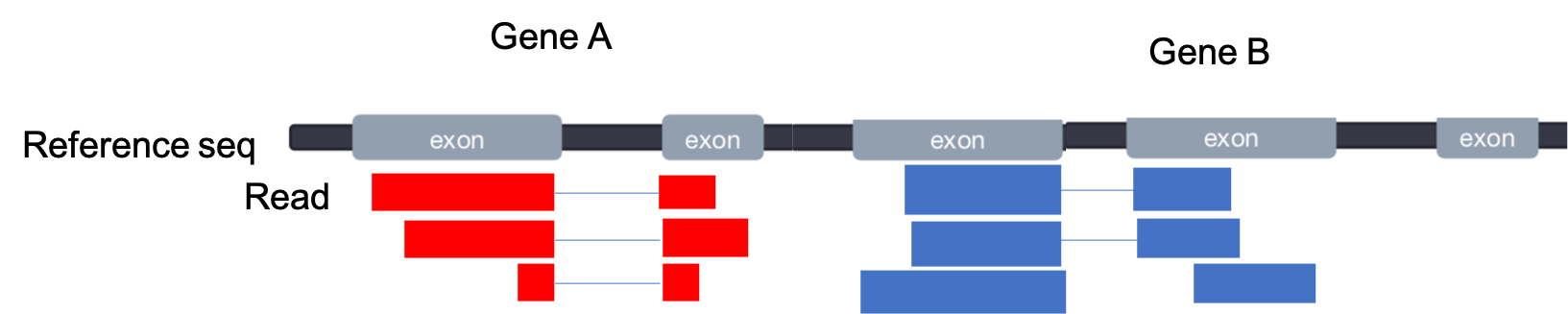
The tool featureCounts is part of the subRead package.
- The mapped coordinates of each read are compared with the features in the GTF file
- Reads that overlap with a gene by >=1 bp are counted as belonging to that feature
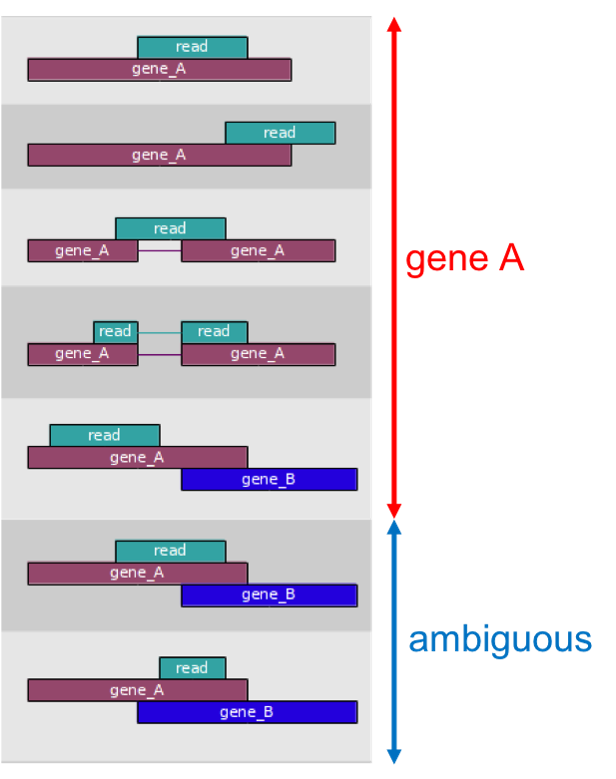
- In default mode, ambiguous reads will be discarded
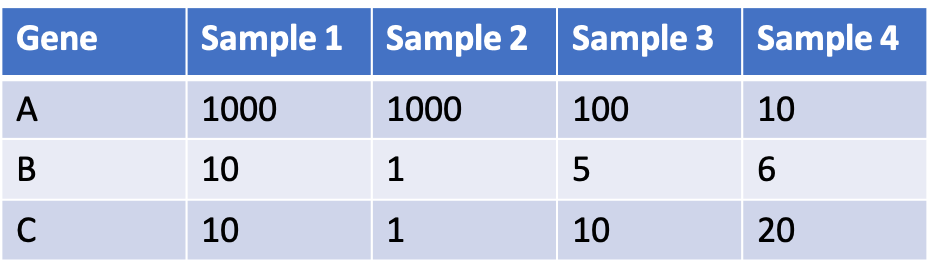
The result is a gene count matrix:
Running featureCounts
- In the Tools panel search bar, type featureCounts
- Select featureCounts under RNA-seq
- Under Alignment file click the
 and select the bam collection generated by STAR
and select the bam collection generated by STAR - Under Gene annotation file select in your history
- Select hg38_genes.gtf
- Click Execute
- The result will be two collections: Summary and Counts
- View the Counts file for a sample by clicking the collection and clicking the

- Run MultiQC on the Summary collection (similar to previous steps, except selecting the appropriate tool (featureCounts) and input folder (featureCounts Summary)).